You've Seen 100 Species. How Many Exist?
April 6, 2026
Given a sample of observations from a discrete distribution, how much remains unseen? The fingerprint – the vector of frequencies of frequencies ( singletons, doubletons, …) – is a sufficient statistic for all symmetric properties of the distribution. Good-Turing uses to estimate unseen mass, Chao1 uses as a lower bound on support size, and the same structure underlies Turing’s wartime cipher work, Shakespeare authorship forensics, and the HyperLogLog sketches in production databases.
In the 1940s, the lepidopterist Alexander Corbet spent two years trapping butterflies in Malaya. He returned with a frequency table: 118 species observed exactly once, 74 observed twice, 44 three times, and so on. He brought the table to Ronald Fisher and asked: if I went back for another two years, how many new species would I find?
Fisher had to estimate a column of the table that didn’t exist: the zero column, the species Corbet never saw. Around the same time, at Bletchley Park, Alan Turing was working on the same problem in a different guise: estimating the probability of encountering a cipher pattern that had never appeared in intercepted traffic.
Both problems have the same structure: a finite sample from a distribution whose support is unknown and possibly much larger than what you’ve observed. The answer depends on the frequency of frequencies: not just how many species you saw, but how many you saw exactly once, exactly twice, exactly three times. This vector is the fingerprint of the sample, and it contains nearly all the information you need.
# The fingerprint
Let be i.i.d. draws from a discrete distribution over an unknown support. Define:
The count is the number of singletons (species seen exactly once), and is the number of doubletons. The vector is the fingerprint.
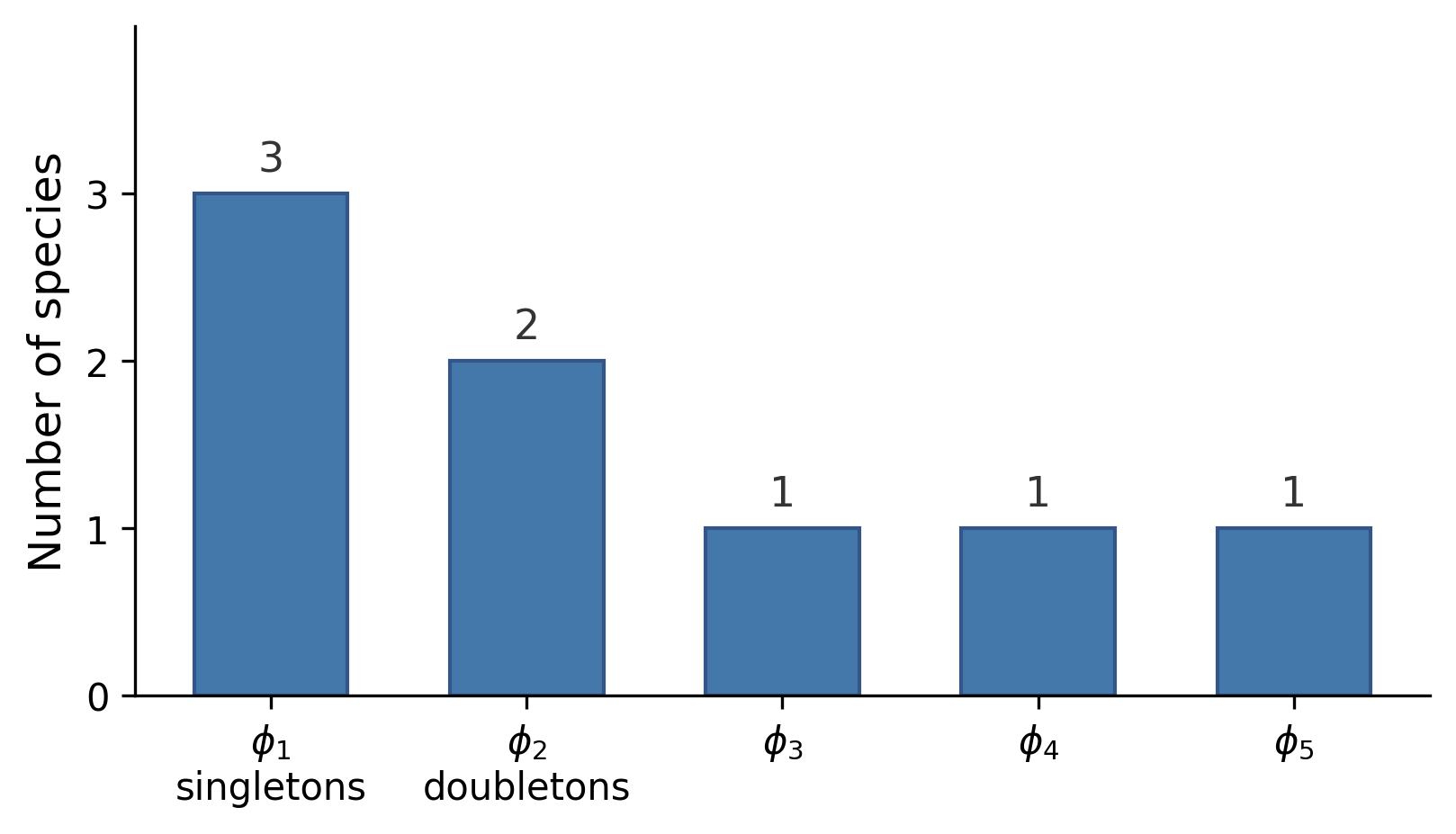
For estimating symmetric properties of a distribution (properties that depend only on the multiset of probabilities, not on which symbol has which probability), the fingerprint is a sufficient statistic. Any estimator based on the full sample can be matched (up to small error) by one using only the fingerprint.1 A practical consequence: you can share a fingerprint without revealing which specific symbols were observed, and any downstream analysis of symmetric properties loses essentially nothing.
The singleton count is large when there are many rare species, species you barely caught. If is large relative to , you’ve probably missed a lot.
The code examples below use fingerprints, a Rust crate named after this data structure.
use fingerprints::Fingerprint;
let counts = [5usize, 4, 3, 2, 2, 1, 1, 1];
let fp = Fingerprint::from_counts(counts).unwrap();
assert_eq!(fp.sample_size(), 19); // n = 5+4+3+2+2+1+1+1
assert_eq!(fp.observed_support(), 8); // S_obs
assert_eq!(fp.singletons(), 3); // phi_1
assert_eq!(fp.doubletons(), 2); // phi_2
# Good-Turing: the probability of the unseen
Good (1953) gave an answer to “what fraction of the next observation will be a species we haven’t seen yet?”2
The missing mass (the total probability of all unseen species) is estimated by the fraction of singletons. If 3 of your 19 observations come from species seen only once, then roughly of the distribution’s probability mass belongs to species you never saw.
Why does this work? Under Poisson sampling, . For well-sampled species (), the factor , so they contribute nothing. For unseen species (), , so they contribute their full probability . The sum is approximately the total probability of species too rare to have appeared: the missing mass. Under Poisson sampling the estimator is exactly unbiased; under multinomial sampling it is approximately unbiased. Either way it uses no assumptions about the shape of .McAllester and Schapire (COLT 2000) give a distribution-free concentration bound of for the missing mass estimator, requiring no distributional assumptions at all.
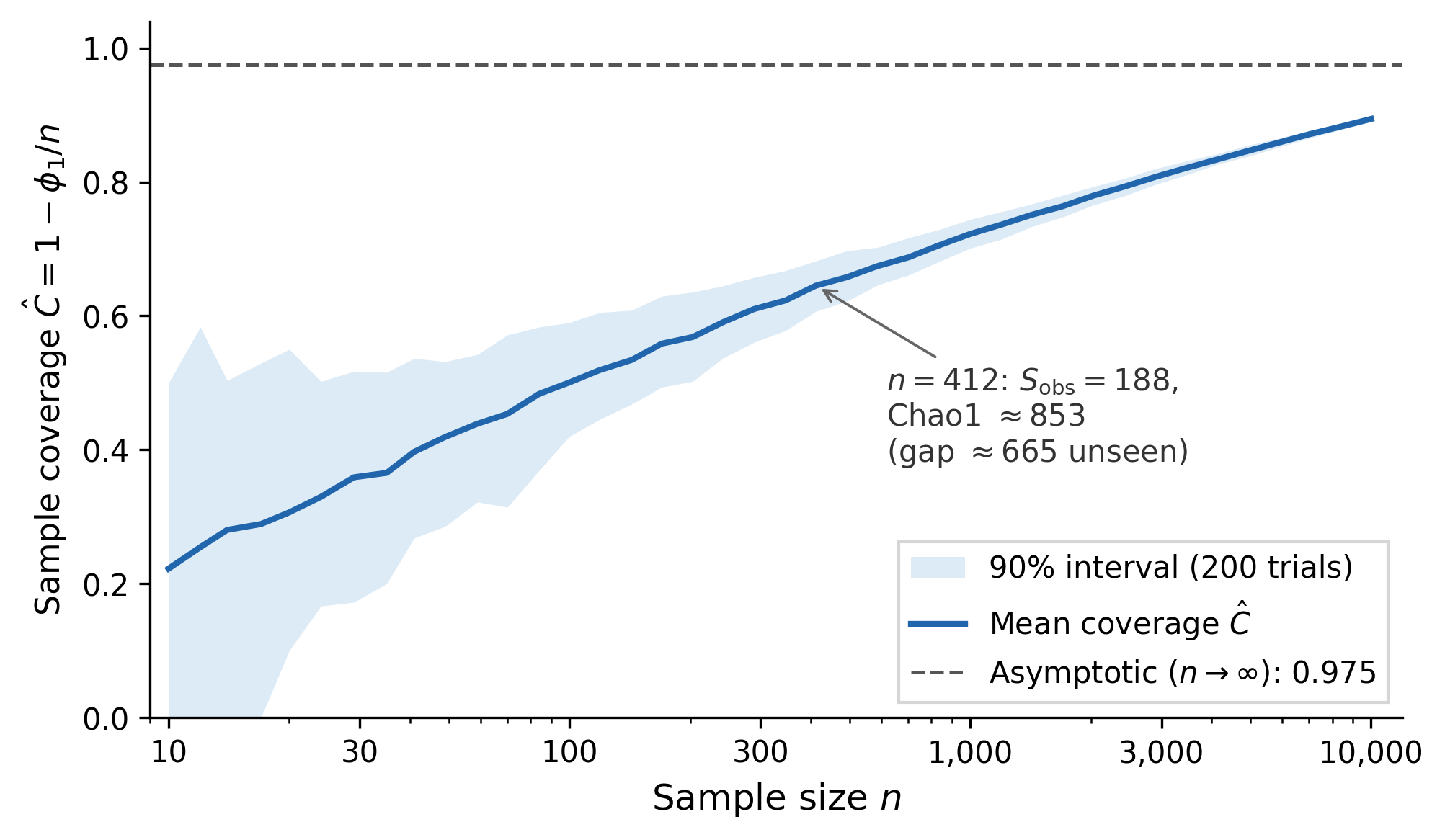
The complementary quantity is sample coverage: . Coverage of 84% means the observed species account for 84% of draws from the underlying distribution.
More generally, Good-Turing smoothing reassigns probabilities to every frequency class: a species seen times gets estimated probability where .17This has a pathology at : the most frequent species gets probability zero because . Gale and Sampson (1995) fix this with log-linear smoothing of the frequency spectrum. The missing mass formula is the special case .
There is a fundamental limit on species-count extrapolation. Orlitsky, Suresh, and Wu (2016) proved that predicting how many new species appear in future samples works when , but beyond that horizon no estimator is consistent.18 This result is about forecasting new-species counts, not about the missing mass estimator itself: for missing mass, the minimax risk is with no phase transition.Rajaraman, Thangaraj, and Suresh (2017, arXiv:1705.05006) establish the minimax lower bound for missing mass estimation, showing risk and no analogue of the log n threshold.
Good formalized and published Turing’s wartime work in 1953.2 The same smoothing technique became the foundation of statistical language modeling. Chen and Goodman’s 1998 survey16 showed that every practical n-gram model before neural methods used Good-Turing smoothing or a direct descendant (Katz backoff, Kneser-Ney).
The Shakespeare test is the most vivid validation: Efron and Thisted (1976) used the fingerprint of word frequencies in Shakespeare’s known canon to predict how many new words would appear if an additional text were discovered.Shakespeare’s known works contain 31,534 distinct word types across roughly 884,000 tokens. Of those, 14,376 appear exactly once – hapax legomena. That alone is 46% of the vocabulary. When an unknown poem attributed to “W.S.” surfaced in the Bodleian Library in 1985, their model predicted new words. The actual count was 9. They also tested poems by Jonson, Marlowe, and Donne against the Shakespearean fingerprint – all failed. The fingerprint became a forensic signature.10
# The minimal-bias extension
Good-Turing uses only , discarding information in . A minimal-bias estimator uses all fingerprint entries:
The alternating signs come from inclusion-exclusion: each term corrects the over- or under-counting of the previous one. The first term is exactly Good-Turing; the remaining terms are its exact bias correction. This has exponentially smaller bias than Good-Turing, at the cost of higher variance for highly skewed distributions.
# How many species total?
# Chao1: a lower bound
Chao (1984) gave a lower bound on the total number of species :3
This is a lower bound: it never overestimates the true support size in expectation. Chao’s original proof applies the Cauchy-Schwarz inequality to , , under binomial sampling; the same form emerges from a Poisson-mixture argument. The inequality is tight only when all unseen species have equal probability; any heterogeneity in abundances means the true is strictly larger. The bound also requires : when the sample is small relative to the square root of the true support, Chao1 can fall substantially short of the true count.Rajaraman, Chandra, Thangaraj, and Suresh (2020, arXiv:2001.04130) characterize convergence rates for Chao1, showing that reliable performance requires . For Zipf(1.1) over types, this threshold is around .
The term estimates the unseen count from the ratio of singletons to doubletons. If you have many singletons but few doubletons, the singletons look like the tip of an iceberg.When , the formula divides by zero. Software implementations use the bias-corrected fallback . This case arises more often than you’d expect – small samples from skewed distributions routinely have zero doubletons.
use fingerprints::{Fingerprint, support_chao1, support_chao1_with_ci};
let fp = Fingerprint::from_counts([5, 3, 2, 1, 1]).unwrap();
let s_hat = support_chao1(&fp);
assert!(s_hat >= fp.observed_support() as f64);
// With variance and 95% CI (log-transformation, Chao 1987):
let est = support_chao1_with_ci(&fp);
println!("[{:.1}, {:.1}, {:.1}]", est.ci_lower, est.point, est.ci_upper);
A cautionary note: Chao1 is a standard alpha-diversity metric in microbiome tools like QIIME2 and mothur, but ASV-based denoising pipelines (ASVs) collapse rare variants, destroying the singleton count that Chao1 depends on. Deng, Umbach, and Neufeld (2024) warned that Chao1 “must not be used” with ASV data13, a case where preprocessing violates the estimator’s assumption. If your pipeline collapses rare types before counting, and no longer mean what Chao1 thinks they mean.
# iChao1: pulling in higher frequencies
Chiu, Wang, Walther, and Chao (2014) improved the lower bound using (tripletons) and (quadrupletons):4
It is always at least as large as Chao1 and uses more of the fingerprint. The correction term captures information about the tail that and alone miss.
# Valiant-Valiant: certified bounds via linear programming
Point estimates are useful until someone asks “how sure are you?”: in regulatory filings, published biodiversity claims, or any setting where overestimation carries a cost. Valiant and Valiant showed that linear programming can extract certified upper and lower bounds on any symmetric property from the fingerprint.5The sample-complexity result is VV STOC 2011 ( optimal rate). The LP-bounding technique implemented in entropy_bounds_lp / support_bounds_lp is from the follow-up VV STOC 2013 “Estimating the Unseen: Improved Estimators.”
The fingerprint from samples constrains the underlying histogram of probabilities. Formulate an LP whose variables are the counts of probability bins, whose constraints require the expected fingerprint to match the observed one (under Poisson sampling), and whose objective minimizes or maximizes the property of interest. This yields certified upper and lower bounds.
The following example shows how wide the bounds can be from 19 observations; the interval itself quantifies uncertainty:
use fingerprints::{Fingerprint, to_bits};
use fingerprints::vv::{support_bounds_lp, entropy_bounds_lp, LpParams};
let fp = Fingerprint::from_counts([5, 4, 3, 2, 2, 1, 1, 1]).unwrap();
let params = LpParams::default_for(&fp);
let (s_lo, s_hi) = support_bounds_lp(&fp, params.clone()).unwrap();
println!("support in [{:.0}, {:.0}]", s_lo, s_hi);
// support in [8, 144]
let (h_lo, h_hi) = entropy_bounds_lp(&fp, params).unwrap();
println!("entropy in [{:.2}, {:.2}] bits", to_bits(h_lo), to_bits(h_hi));
// entropy in [0.16, 4.92] bits
# Entropy in the unseen regime
Estimating Shannon entropy from a finite sample is biased. The naive plug-in estimator systematically underestimates because it misses unseen species entirely.
Four estimators form a bias-correction hierarchy.
# 1. Plug-in (maximum likelihood)
Negatively biased. Appropriate when .
# 2. Miller-Madow
A first-order bias correction due to Miller (1955).6 Simple, but when (many singletons), the correction can overshoot.
# 3. Jackknife
Delete-one jackknifing reduces bias to :7
Computed efficiently from the fingerprint without enumerating all leave-one-out samples.
These four estimators do not exhaust the field. Jiao, Venkat, Han, and Weissman (2015) and Wu and Yang (2016) showed that polynomial approximation methods achieve the minimax-optimal rate for entropy estimation – the same rate VV established – with tighter constants.19 Hao and Orlitsky (NeurIPS 2019) proved that Profile Maximum Likelihood is simultaneously sample-optimal for every 1-Lipschitz symmetric property (entropy, support size, coverage, Rényi divergence) without knowing which property you want in advance.20 These are the right tools in the regime where is within a constant factor of .
# 4. Pitman-Yor
Archer, Park, and Pillow (2012, 2014) showed that the Pitman-Yor process, a two-parameter generalization of the Dirichlet process, is a better prior for entropy estimation because it generates power-law tails.8 The process has discount and concentration . When , the number of distinct symbols grows as rather than for the Dirichlet process (), making it appropriate for natural language, species distributions, and network degree distributions. A related Bayesian approach is the NSB estimator of Nemenman, Shafee, and Bialek (2001), standard in neuroscience, which places a prior approximately uniform over entropy values – a practically useful invariance when the support size is unknown.21
use fingerprints::{Fingerprint, entropy_pitman_yor_nats, pitman_yor_params_hat, to_bits};
let fp = Fingerprint::from_counts([5, 4, 3, 2, 2, 1, 1, 1]).unwrap();
let h_py = entropy_pitman_yor_nats(&fp);
let py = pitman_yor_params_hat(&fp);
println!("H = {:.3} bits (d={:.3}, alpha={:.3})",
to_bits(h_py), py.d, py.alpha);
# A worked example
Sampling from a Zipf$(s=1.1)$ distribution over types, then measuring each estimator’s error in bits relative to the true . Columns: Plug-in, Miller-Madow, Jackknife, Pitman-Yor.
N S F1 p0_hat err_plug err_MM err_JK err_PY
50 34 26 0.5200 -3.1185 -2.6424 -2.2469 -1.2323
100 54 41 0.4100 -2.8684 -2.4861 -2.1760 -1.0118
200 101 82 0.4100 -2.2451 -1.8845 -1.5798 -0.2188
400 192 148 0.3700 -1.3858 -1.0413 -0.7629 +0.5478
800 299 228 0.2850 -1.4200 -1.1513 -0.9360 +0.5788
1600 510 382 0.2387 -1.0018 -0.7723 -0.5924 +0.9947
3200 847 576 0.1800 -0.6455 -0.4548 -0.3162 +1.2493
6400 1350 857 0.1339 -0.4401 -0.2880 -0.1835 +1.3877
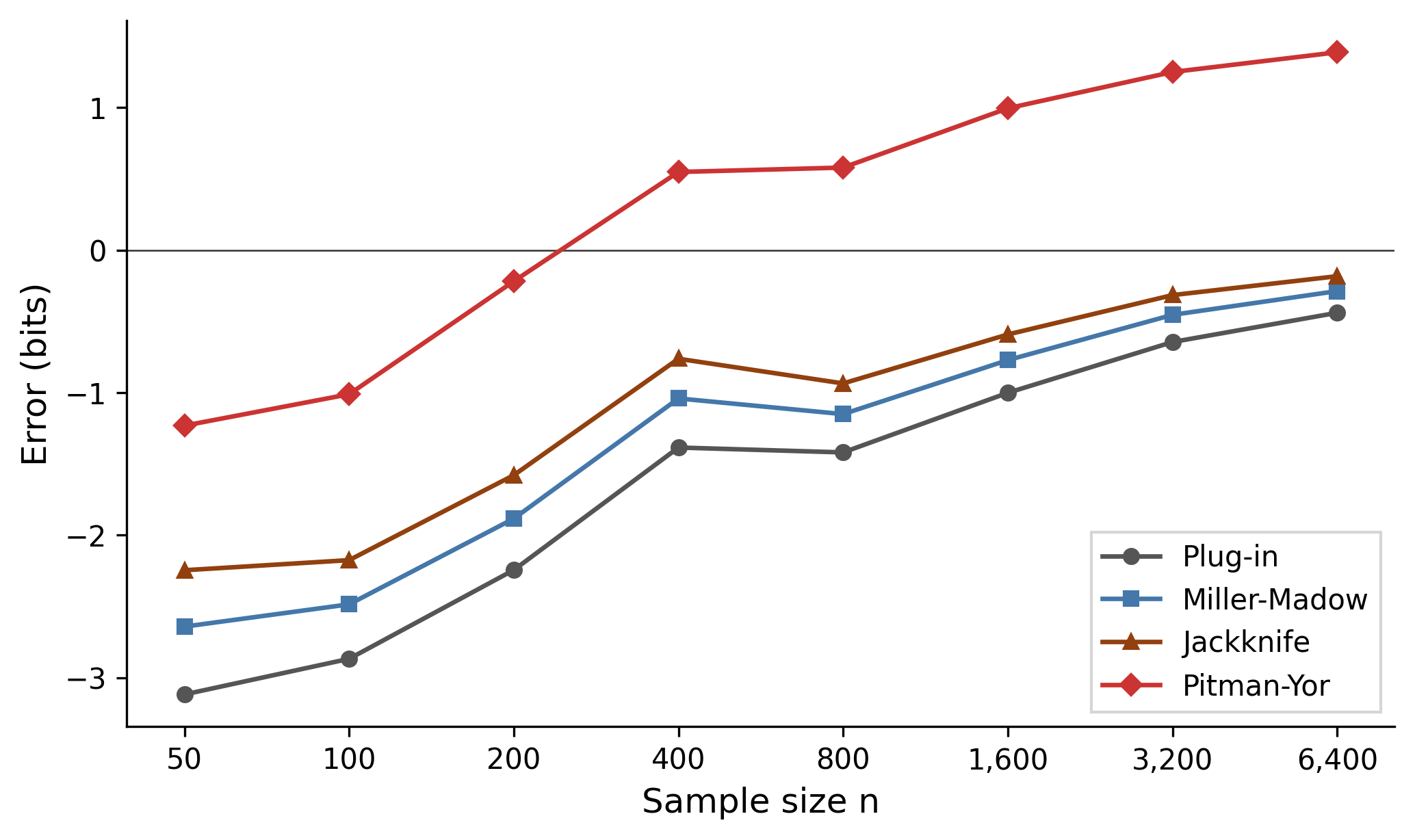
At , coverage is only 48% and the plug-in underestimates by 3.1 bits. The Pitman-Yor estimator has the smallest absolute error at small sample sizes: at , it’s off by just 0.2 bits while the plug-in is off by 2.2.
But as grows, PY overshoots: by it overestimates by 1.4 bits while the jackknife is off by only 0.2. The reason is visible in the fitted parameters: the discount across all sample sizes, meaning the MLE cannot distinguish this Zipf tail from geometric decay.This is a finite-sample MLE artifact. Asymptotic theory predicts for Zipf(); the MLE needs far larger than this experiment uses to recover that value. The pathology is real but particular to small samples, not a fundamental indistinguishability of Zipf from geometric.
With , the Pitman-Yor process reduces to a Dirichlet process, a simpler model whose vocabulary grows as rather than the power-law that a Zipf distribution actually generates. The model sees more singletons than an vocabulary can explain, misattributes them to a vast unseen population, and overcompensates for missing mass the data has already revealed. The classical estimators are boring but reliable: consistently negatively biased, converging steadily toward zero.
# When estimators disagree
Different estimators make different assumptions about the tail. When they converge to similar values, the tail assumption doesn’t matter. When they diverge, the divergence tells you the sample is too small to resolve the tail, and no estimator can fix that.
| Regime | Recommended estimator |
|---|---|
| , few singletons | Plug-in or Miller-Madow |
| Moderate undersampling | Jackknife |
| Severe undersampling, power-law tails | Pitman-Yor or NSB |
| , minimax-optimal for entropy | JVHW polynomial or PML |
| Need certified bounds, not point estimates | Valiant-Valiant LP |
| Need lower bound on support size | Chao1 or iChao1 |
When sample coverage (i.e., ), plug-in and Miller-Madow should be treated with caution. At very small , Pitman-Yor produces a less biased estimate, but as the convergence plot shows, it can overshoot at larger sample sizes. No single estimator dominates across all regimes. The safest strategy: report multiple estimates and the VV bounds, letting the spread quantify your uncertainty.
Two applications make the stakes concrete. In forensic DNA, when a suspect’s DNA profile matches a crime scene sample but has never appeared in the reference database, classical frequency estimation gives a match probability of zero. Absurd. This is the missing mass problem directly: “what is the probability of a type we haven’t seen?” Favaro and Naulet (2023) frame it as a Good-Turing problem and prove optimal bounds.12
In software fuzzing, Böhme (2018) mapped the framework onto security testing: execution paths are species, fuzzer runs are samples. Good-Turing estimates how many undiscovered paths remain: the residual risk that a vulnerability lurks in uncovered code.14 When your estimators disagree on how many paths remain, you haven’t fuzzed enough.
# The fingerprint everywhere
The same structure recurs far beyond ecology.
Your immune system contains distinct T-cell receptor clonotypes generated by VDJ recombination, but any blood draw captures a tiny fraction. Each clonotype is a species, each sequenced cell is a sample. Laydon et al. (2015) showed that classical estimators severely undercount because the TCR frequency distribution is heavy-tailed.11 An unusually diverse repertoire correlates with effective viral control, and the unseen fraction has direct clinical relevance for HIV, cancer immunotherapy, and aging.
At the other end of the scale, HyperLogLog (Flajolet et al., 2007) estimates the number of distinct elements in a data stream – the same “how many species?” question, solved with hash-based sketches instead of fingerprints.The connection goes deeper than analogy. Cohen (2016) extended HyperLogLog to estimate the full frequency-of-frequencies structure, explicitly bridging the database and ecological traditions. It’s deployed in Redis, Spark, Google’s infrastructure, and PostgreSQL via an extension. The question Fisher and Good posed for butterfly species in the 1940s is now answered billions of times per day in production databases.15
Even the German tank problem fits: Allied statisticians estimated German tank production from captured serial numbers. The MVUE (where is the max serial, is sample size) gave estimates within 1% of actual production. This is the uniform-distribution special case. The underlying question is the same – estimating a population from a sample – though the mathematical tools differ.
The unseen species problem is one of those places where a single mathematical structure, the fingerprint, connects Bletchley Park to microbiome sequencing to forensic evidence to database cardinality. The question is always the same: you’ve seen some of it. How much is left?
# References
[1] Acharya, Das, Orlitsky, Suresh, “A unified maximum likelihood approach for estimating symmetric properties of discrete distributions,” ICML 2017. ↩
[2] I.J. Good, “The population frequencies of species and the estimation of population parameters,” Biometrika 40(3/4), 237–264, 1953. The core idea is due to Turing; Good credits him explicitly but is the sole listed author. ↩
[3] Anne Chao, “Nonparametric estimation of the number of classes in a population,” Scandinavian Journal of Statistics 11(4), 265–270, 1984. ↩
[4] Chiu, Wang, Walther, Chao, “An improved nonparametric lower bound of species richness via a modified Good-Turing frequency formula,” Biometrics 70(3), 671–682, 2014. ↩
[5] Gregory Valiant, Paul Valiant, “Estimating the unseen: An -sample estimator for entropy and support size, shown optimal via new CLTs,” STOC, 685–694, 2011. Journal version: JACM 64(6), 2017. The LP-bounding approach is from the follow-up: “Estimating the Unseen: Improved Estimators for Entropy and other Properties,” STOC 2013. ↩
[6] G.A. Miller, “Note on the bias of information estimates,” in Information Theory in Psychology, Free Press, 95–100, 1955. Often called “Miller-Madow” by convention, though Madow is not a co-author. ↩
[7] S. Zahl, “Jackknifing an index of diversity,” Ecology 58(4), 907–913, 1977. ↩
[8] Evan Archer, Il Memming Park, Jonathan W. Pillow, “Bayesian entropy estimation for countable discrete distributions,” JMLR 15, 2833–2868, 2014. Conference version: NeurIPS 2012. ↩
[10] Bradley Efron, Ronald Thisted, “Estimating the number of unseen species: How many words did Shakespeare know?”, Biometrika 63(3), 435–447, 1976. The validation: Thisted and Efron, “Did Shakespeare write a newly-discovered poem?”, Biometrika 74(3), 445–455, 1987. ↩
[11] Laydon, Bangham, et al., “Estimating T-cell repertoire diversity: limitations of classical estimators and a new approach,” Phil. Trans. R. Soc. B 370, 20140291, 2015. ↩
[12] Favaro, Naulet, “Optimal estimation of high-order missing masses, and the rare-type match problem,” arXiv:2306.14998, 2023. ↩
[13] Deng, Umbach, Neufeld, “Nonparametric richness estimators Chao1 and ACE must not be used with amplicon sequence variant data,” The ISME Journal, 2024. ↩
[14] Marcel Böhme, “STADS: Software Testing as Species Discovery,” ACM TOSEM 27(2), 2018. ↩
[15] Flajolet, Fusy, Gandouet, Meunier, “HyperLogLog: the analysis of a near-optimal cardinality estimation algorithm,” AofA, 2007. Cohen, “HyperLogLog Hyper Extended,” arXiv:1607.06517, 2016. ↩
[16] Chen, S. F. & Goodman, J. (1998). “An empirical study of smoothing techniques for language modeling.” Technical Report TR-10-98, Harvard University. ↩
[17] Gale, W. A. & Sampson, G. (1995). “Good-Turing frequency estimation without tears.” Journal of Quantitative Linguistics, 2(3), 217–237. ↩
[18] Orlitsky, A., Suresh, A. T. & Wu, Y. (2016). “Optimal prediction of the number of unseen species.” Proceedings of the National Academy of Sciences, 113(47), 13283–13288. ↩
[19] Jiao, J., Venkat, K., Han, Y. & Weissman, T. (2015). “Minimax estimation of functionals of discrete distributions.” IEEE Transactions on Information Theory, 61(5), 2835–2885. arXiv:1406.6956. Wu & Yang (2016, arXiv:1407.0381) give tight constants for entropy. ↩
[20] Hao, Y. & Orlitsky, A. (2019). “Unified sample-optimal property estimation in near-linear time.” NeurIPS 2019. arXiv:1906.03794. ↩
[21] Nemenman, I., Shafee, F. & Bialek, W. (2001). “Entropy and inference, revisited.” NeurIPS 2001. arXiv:physics/0108025. ↩